Metal Docking in PrinS³: Expanding Ligand Binding Studies to Metal-Dependent Systems
Metallo-β-lactamases (MBLs) are a critical group of enzymes that enable bacteria to resist β-lactam antibiotics, including carbapenems, which are often considered the last line of defense for treating severe bacterial infections. Carbapenems, a powerful subclass of β-lactam antibiotics, are commonly used to treat serious infections caused by multidrug-resistant (MDR) bacteria due to their broad-spectrum activity against both Gram-positive and Gram-negative pathogens. These drugs are typically reserved for cases where other antibiotics have proven ineffective. MBLs are predominantly found in Gram-negative bacteria such as Klebsiella pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii. These pathogens frequently exhibit multidrug resistance, making treatment more challenging. Furthermore, MBLs are zinc-dependent enzymes that hydrolyze the β-lactam ring of antibiotics, rendering them ineffective and contributing to antibiotic resistance. The metal ions, typically zinc, play a crucial role in the catalytic mechanism and structural integrity of these enzymes. The essential role of zinc ions in MBL mechanisms makes them a target for inhibitor design. Chelating agents or compounds that disrupt zinc coordination could potentially inhibit MBL activity and restore antibiotic efficacy. To combat this resistance, researchers are prioritizing the development of combination therapies and novel inhibitors.
The PrinS³ platform, already robust for small-molecule docking, molecular dynamics, and MM-PBSA workflows, can be enhanced with metal docking capabilities. Metal-dependent enzymes and complexes, such as metallo-β-lactamases (MBLs) or metalloproteins, require specialized approaches for accurate simulation and prediction of ligand binding.
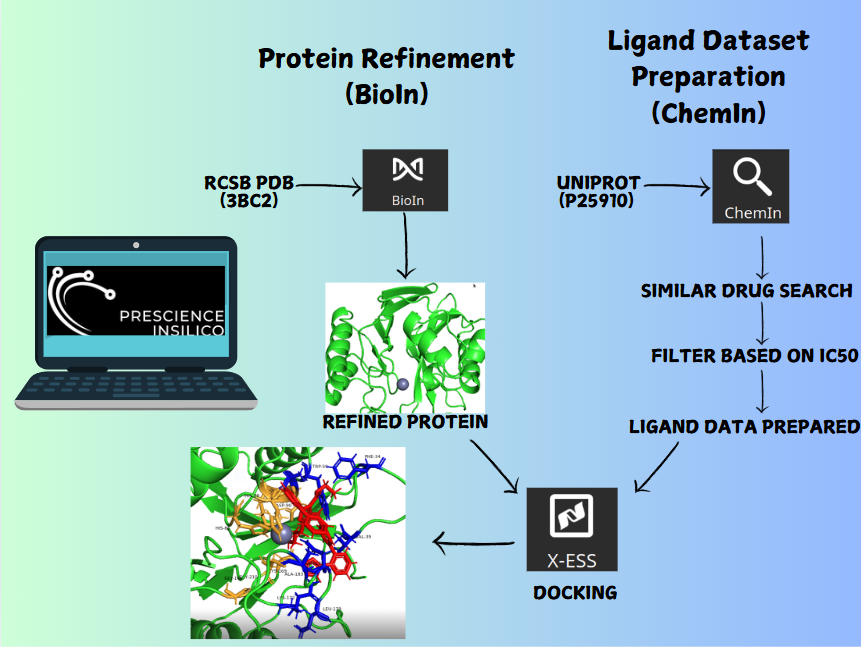
The following workflow outlines the step-by-step approach to identifying potential drug candidates targeting Metallo Beta-Lactamase II from Bacillus cereus (UniProt ID: P25910) using PrinS³ tools.
1. Dataset Generation: Create a dataset of small molecules with potential as drug candidates.
- Ligand Selection:
- Identified the target protein using its UniProt ID (P25910).
- Conducted a search in the UniProt database for chemically similar compounds using ChemIn, ensuring relevance to the target.
- Compound Filtering:
- Selected compounds with IC₅₀ values ≤ 20,000 for optimal activity.
- Converted the selected compounds into PDB format for molecular docking studies.
2. Protein Preparation: Ensure the structural accuracy of the target protein for docking studies.
- Protein Structure Retrieval:
- Downloaded the crystal structure of Metallo Beta-Lactamase II from the RCSB Protein Data Bank.
- Structure Refinement:
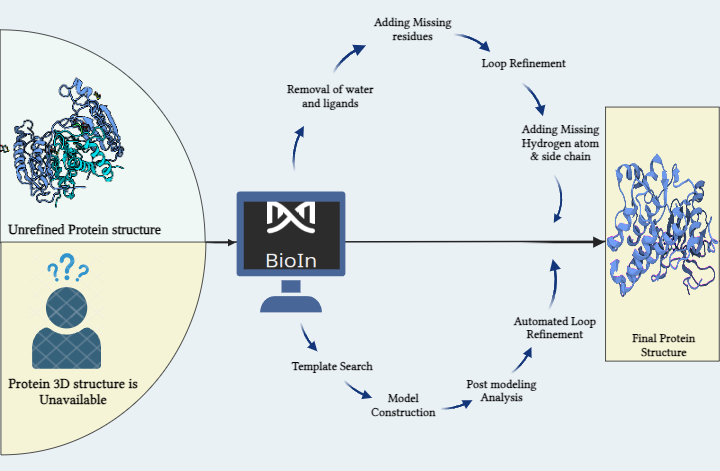
- Utilized the BioIn tool in PrinS³ to refine the protein structure for computational studies.
- This step optimized bond angles, removed unnecessary ligands, and added missing residues.
3. Molecular Docking: Screening binding interactions between the target protein and ligand dataset.
- Docking Setup:
- Used the X-ESS tool in PrinS³ for molecular docking.
- Identified active sites through meta-analysis of structural and functional data.
- Created a grid box at the docking site to ensure ligand accessibility.
- Validation:
- Visualized the grid box placement to confirm accuracy before executing the docking process.
4. Interaction Analysis: Identify lead compounds with strong binding affinity and favorable interactions.
- Analysis Criteria:
- Evaluated docking results based on minimum binding energy values.
- Highlighted one compound that demonstrated numerous interactions analogous to the original inhibitor.
- Key interactions included hydrogen bonds, π-stacking, and metal coordination with the active site residues.
This workflow enabled the identification of a promising lead compound with significant binding affinity to Metallo Beta-Lactamase II. The integrated tools within PrinS³, such as ChemIn, BioIn, and X-ESS, streamlined the ligand selection, protein preparation, and docking analysis, enhancing the efficiency of the drug discovery process.
For further details, reach out to us at support@prescience.in